COMUNICACIÓN POSTER
AUTORES
Velilla Alonso, Gabriel; Palacios Mendoza, Michael Armando; Guillem Mesado, Maria Desamparados; Saldaña Díaz, Ana Isabel
CENTROS
Servicio de Neurología. Hospital General Universitario Gregorio Marañón
OBJETIVOS
Describir las características clínicas de los casos de síndrome de cefalea y déficit neurológico transitorio con pleocitosis linfocitaria en líquido cefalorraquídeo (HaNDL).
MATERIAL Y MÉTODOS
Se recogieron retrospectivamente datos demográficos, clínicos y pruebas complementarias de los pacientes diagnosticados de síndrome de HaNDL en nuestro centro entre Enero/2015-Enero/2020.
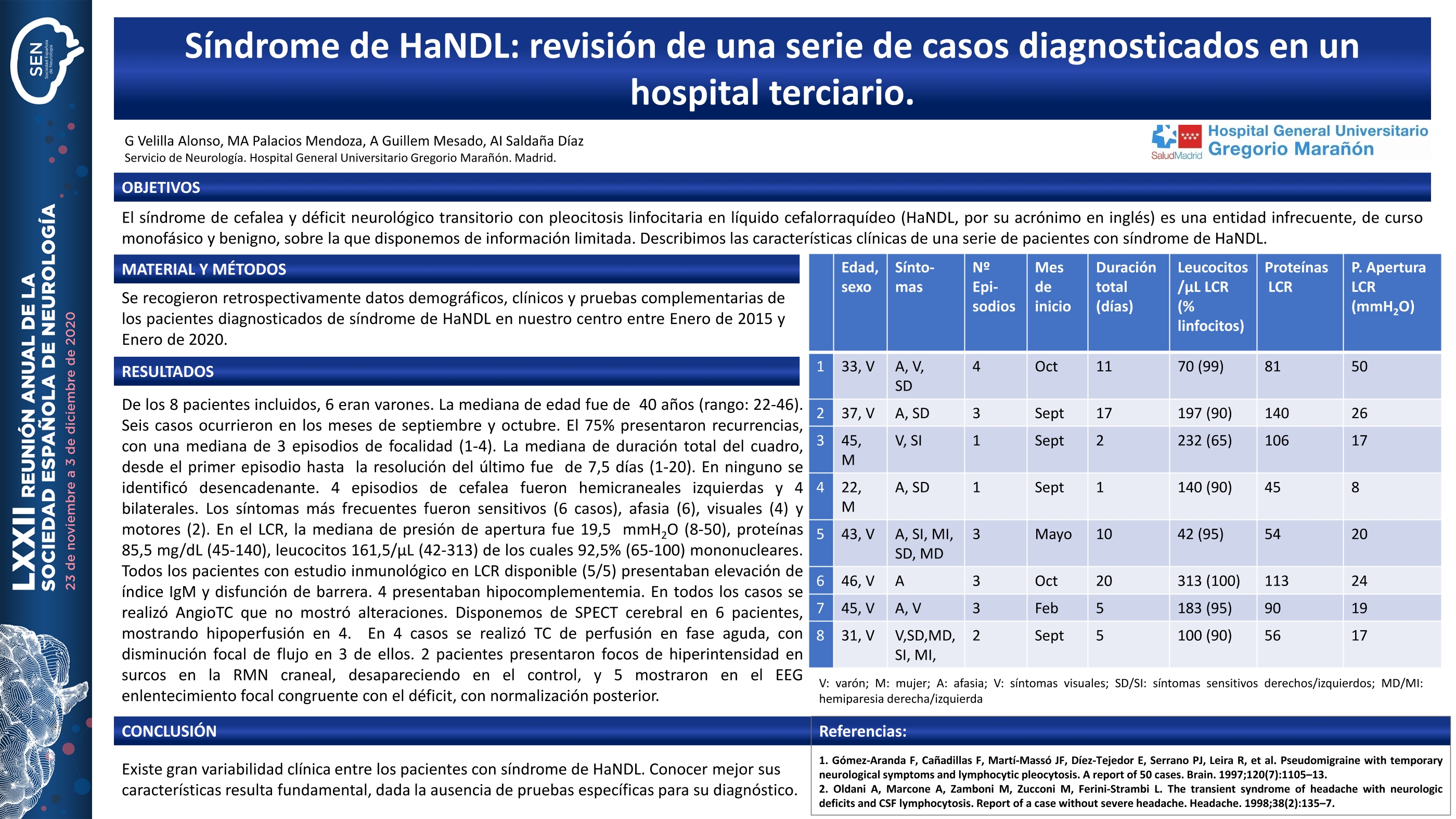
RESULTADOS
De los 8 pacientes incluidos, 6 eran varones. La mediana de edad fue de 40 años (rango:22-46). Seis casos ocurrieron en los meses de septiembre/octubre. El 75% presentaron recurrencias, mediana de 3 episodios de focalidad (1-4). La mediana de duración desde el primer episodio hasta resolución del último fue 7,5 días (1-20). En ninguno se identificó desencadenante. 4 episodios de cefalea fueron hemicraneales izquierdas y 4 bilaterales. Los síntomas más frecuentes fueron sensitivos (6 casos), afasia (6), visuales (4) y motores (2). En LCR, la mediana de presión de apertura fue 19,5 mmHg(8-50), proteínas 85,5 mg/dL(45-140), leucocitos 161,5/μL(42-313), 92,5%(65-100) mononucleares. Todos (5/5) los pacientes con estudio inmunológico en LCR presentaban elevación de índice IgM y disfunción de barrera. 4 presentaban hipocomplementemia. Disponemos de SPECT en 6 pacientes, mostrando hipoperfusión en 4. En 4 se realizó TC de perfusión en fase aguda, con disminución focal de flujo en 3 casos. 2 pacientes presentaron focos de hiperintensidad en surcos en la RMN craneal, desapareciendo en el control, y 5 mostraron en el EEG enlentecimiento focal congruente con el déficit, con normalización posterior.
CONCLUSIONES
Existe gran variabilidad clínica entre los pacientes con síndrome de HaNDL. Conocer mejor sus características es fundamental, dada la ausencia de pruebas específicas para su diagnóstico.