COMUNICACIÓN POSTER
AUTORES
Abril- Jaramillo, Javier 1; Móndejar , R 2; García Moreno, JM 2; Domínguez Mayoral, Ana 2; De Torres Chacón, Reyes 2
CENTROS
1. Trastornos del Movimiento. Servicio de Neurología. Centro de Neurología Avanzada; 2. Servicio de Neurología. Complejo Hospitalario Regional Virgen Macarena
OBJETIVOS
Descripción de un caso clínico de lipoidoproteinosis, genodermatosis de herencia autosómica recesiva poco conocida y poco frecuente
MATERIAL Y MÉTODOS
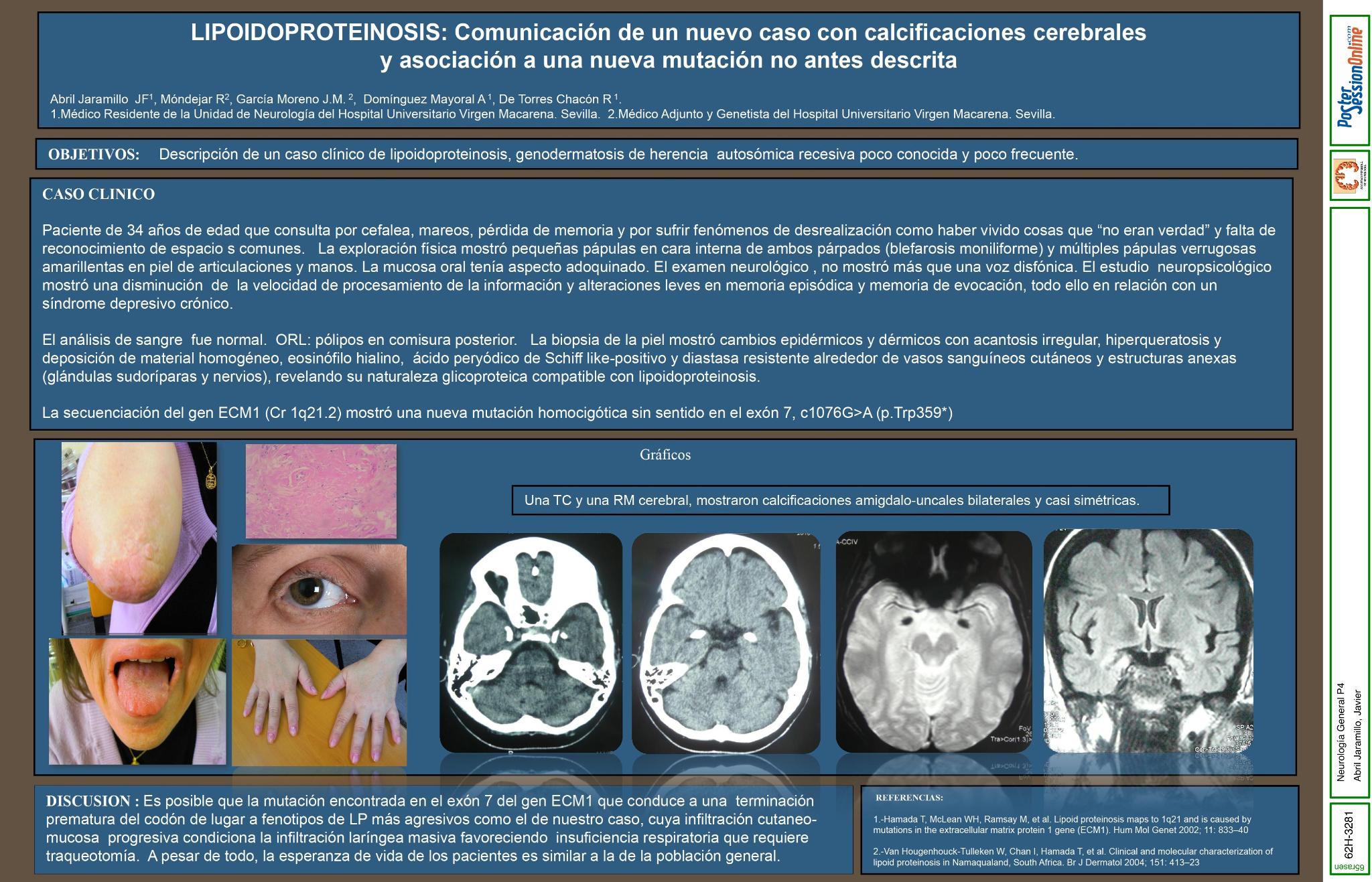
Paciente de 34 años de edad que consulta por cefalea, mareos, pérdida de memoria y por sufrir fenómenos de desrealización como haber vivido cosas que “no eran verdad” y no reconocimiento de lugares comunes. La exploración física mostró pequeñas pápulas en cara interna de ambos párpados (blefarosis moniliforme) y múltiples pápulas verrugosas amarillentas en piel de articulaciones y manos. La mucosa oral tenía aspecto adoquinado. El examen neurológico no mostró más que una voz disfónica. El neuropsicológico mostró una disminución en la velocidad de procesamiento de la información y alteraciones leves en memoria episódica y memoria de evocación, todo ello en relación con un síndrome depresivo crónico
RESULTADOS
El análisis de sangre fue normal. Una TC y una RM cerebral, mostraron calcificaciones amigdalo-uncales bilaterales y casi simétricas. La biopsia de la piel mostró cambios epidérmicos y dérmicos con acantosis irregular, hiperqueratosis y deposición de material homogéneo, eosinófilo hialino ácido peryódico de Schiff like-positivo y diastasa resistente alrededor de vasos sanguíneos cutáneos y estructuras anexas (glándulas sudoríparas y nervios), revelando su naturaleza glicoproteica compatible con lipoidoproteinosis. La secuenciación del gen ECM1 (Cr 1q21.2) mostró una nueva mutación homozigótica sin sentido en el exón 7, c1076G>A (p.Trp359*)
CONCLUSIONES
Es posible que la mutación encontrada en el exón 7 del gen ECM1 que conduce a una terminación prematura del codón de lugar a fenotipos de LP más agresivos como el de nuestro caso