COMUNICACIÓN POSTER
AUTORES
Monterde Ortega, Angela 1; Blanco Valero, Maria del Carmen 2; Rodríguez Martín, Alba 2; Piñar Morales, Raquel 2; Peláez Viña, Nazaret 2
CENTROS
1. Servicio de Neurología. Hospital Universitari Joan XXIII de Tarragona; 2. Servicio de Neurología. Hospital Reina Sofía
OBJETIVOS
El insomnio familiar fatal (IFF) es una enfermedad priónica hereditaria caracterizada por pérdida progresiva del sueño profundo,disautonomía y trastornos motores,posteriormente puede haber diplopia,disartria,disfagia y ataxia.Suele presentarse sobre los 48 años de media.Se produce por una mutación en el codon 178 de la proteína priónica.No tiene tratamiento y es mortal.
MATERIAL Y MÉTODOS
Mujer de 21 años que comienza con diplopia binocular horizontal fluctuante.Se pensó en Miastenia tratándose con mestinon y mejorando parcialmente el cuadro.A los tres meses se añade sensación de inestabilidad,necesitando al mes un apoyo para deambular. A los ocho meses del inicio en la exploración destaca bradipsiquia, voz monótona, fallos en memoria,alucinaciones visuales,paresia de ambos rectos externos,sacadas hipermétricas multidireccionales con ambos ojos,marcha atáxica espástica,hipereplexia, sueño fragmentado,poco reparador y alucinaciones oníricas.
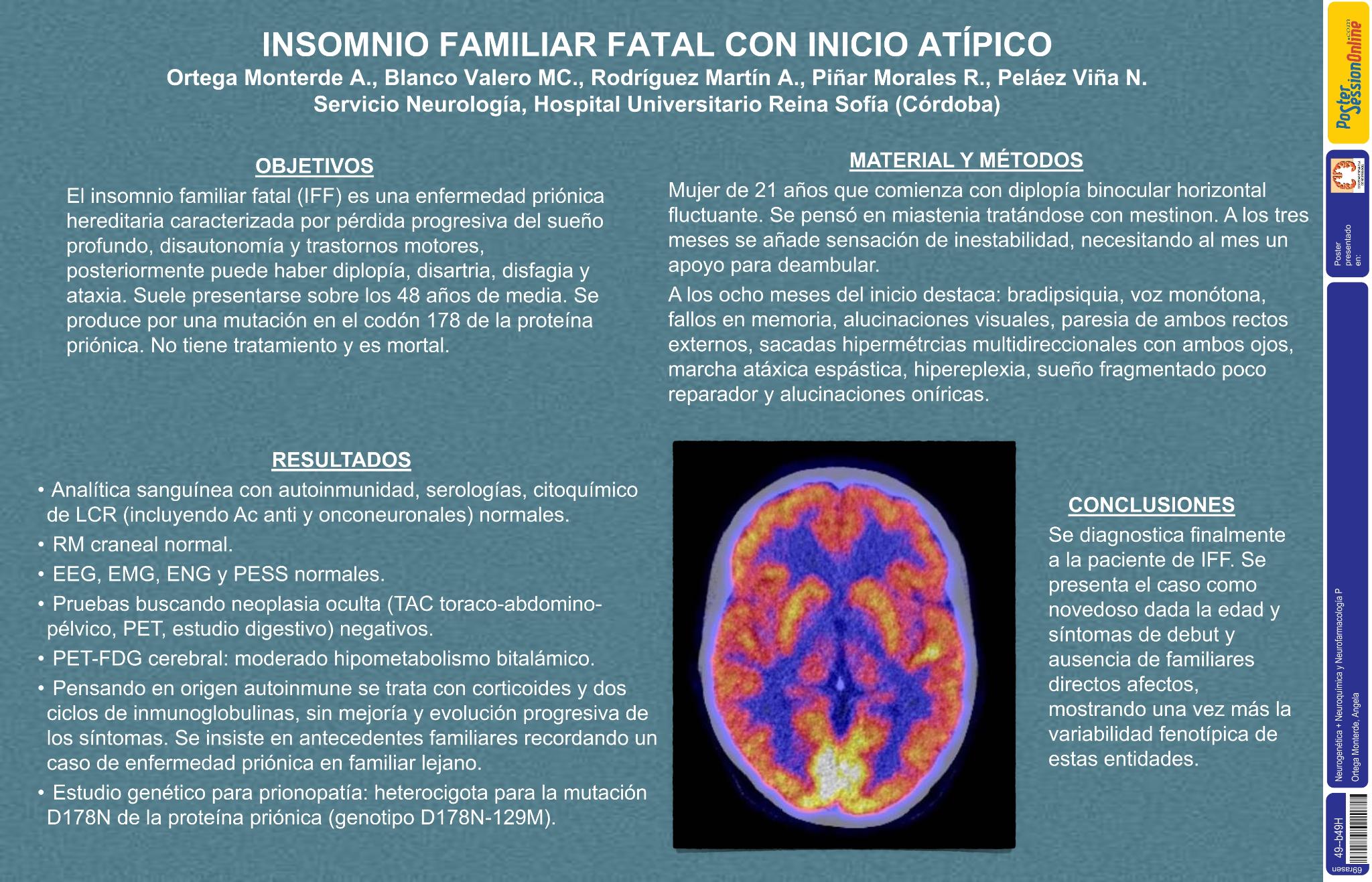
RESULTADOS
Analítica sanguinea incluyendo autoinmunidad,serologías,citoquímico de LCR (incluyendo Ac anti y onconeuronales) normales. RM craneal normal. EEG seriados, EMG, ENG y PESS normales. Pruebas buscando neoplasia oculta (TAC toraco-abdomino-pelvico,PET,estudio digestivo) negativos. Pensando en origen autoinmune se trata con corticoides y dos ciclos de inmunoglobulinas,sin mejoría y evolución progresiva de los síntomas. Se insiste en antecedentes familiares recordando un caso de enfermedad priónica en familiar lejano. PET-FDG cerebral:hipometabolismo bitalámico. Estudio genético para prionopatía: heterocigota para la mutación D178N de la proeína priónica (genotipo D178N-129M).
CONCLUSIONES
Se diagnostica finalmente a la paciente de IFF.Se presenta el caso como novedoso dada la edad y síntomas de debut y ausencia de familiares directos afectos,mostrando una vez más la variabilidad fenotipica de estas entidades.