COMUNICACIÓN POSTER
AUTORES
Cobo Roldán, Lourdes; Peláez Viña, Nazaret; Gómez Caravaca, Maria Teresa ; Blanco Valero, Maria Carmen
CENTROS
Servicio de Neurología. Hospital Reina Sofía
OBJETIVOS
La enfermedad de Pompe (EP) es un trastorno de almacenamiento de glucógeno autosómico recesivo causado por una deficiencia de la enzima α-glucosidasa, lo que conduce a su acumulación en los tejidos, principalmente en el músculo esquelético. Se ha descrito una alteración en la microvasculatura cerebral en las formas de inicio tardío que conducen al sangrado cerebral.
MATERIAL Y MÉTODOS
Varón de 75 años con diagnóstico de enfermedad de Pompe en 2017 tras historia de 9 años de debilidad proximal en miembros inferiores, bajo tratamiento con terapia enzimática sustitutiva desde entonces. Como antecedentes personales, hipertenso, lues latente tardía, ictus isquémico en territorio vertebrobasilar en 2002 con hallazgo de dolicoectasia basilar e infarto de arteria central de la retina de ojo derecho. Ingresa por déficit motor braquiocrural izquierdo de instauración brusca. En la exploración física destaca disartria y hemiparesia izquierda de predominio distal (4/5).
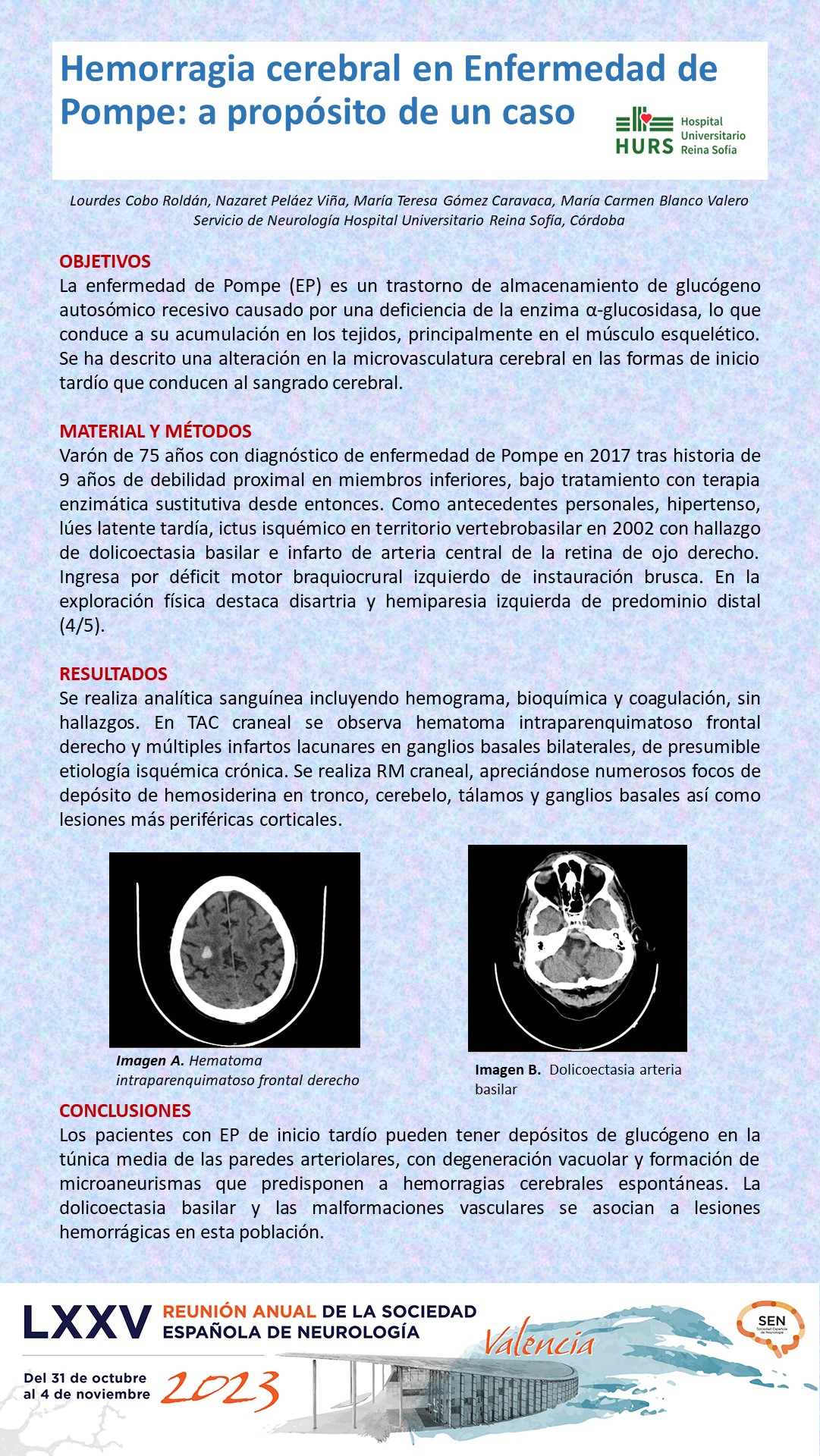
RESULTADOS
Se realiza analítica sanguínea incluyendo hemograma, bioquímica y coagulación, sin hallazgos. En TAC craneal se observa hematoma intraparenquimatoso frontal derecho y múltiples infartos lacunares en ganglios basales bilaterales, de presumible etiología isquémica crónica. Se realiza RM craneal, apreciándose numerosos focos de depósito de hemosiderina en tronco, cerebelo, tálamos y ganglios basales así como lesiones más periféricas corticales.
CONCLUSIONES
Los pacientes con EP de inicio tardío pueden tener depósitos de glucógeno en la túnica media de las paredes arteriolares, con degeneración vacuolar y formación de microaneurismas que predisponen a hemorragias cerebrales espontáneas. La dolicoectasia basilar y las malformaciones vasculares se asocian a lesiones hemorrágicas en esta población.