COMUNICACIÓN POSTER
AUTORES
Ortega Hiraldo, Carmen 1; Carbonell Corvillo, Pilar 2; Rivas Infante, Eloy 3; Romero Godoy, Jorge 2; Gómez González, Ana 2; Aguilar Monge, Alba 2
CENTROS
1. Servicio de Neurología. Hospital Virgen de la Victoria; 2. Servicio de Neurología. Complejo Hospitalario Virgen de la Victoria; 3. Servicio: Anatomía Patológica. Complejo Hospitalario Regional Virgen del Rocío
OBJETIVOS
Las mutaciones en el gen GFPT1 determinan una alteración en el mecanismo de glicosilación del receptor de acetilcolina, produciendo un síndrome miasténico congénito (SMC) que se caracteriza por una debilidad de cinturas, con escasa afectación facial, ocular y bulbar. Nuestro objetivo es describir el caso de dos hermanos con un SMC recesivo asociado a mutaciones en el gen GFPT1.
MATERIAL Y MÉTODOS
Dos pacientes (56 y 53 años) presentaron un cuadro clínico superponible. Refirieron peor condición física en infancia. A partir de los 20 años empezaron con dificultad para subir escaleras que empeoró de forma progresiva, precisando bastón para deambular después delos 40 años. Nunca refirieron síntomas bulbares.
RESULTADOS
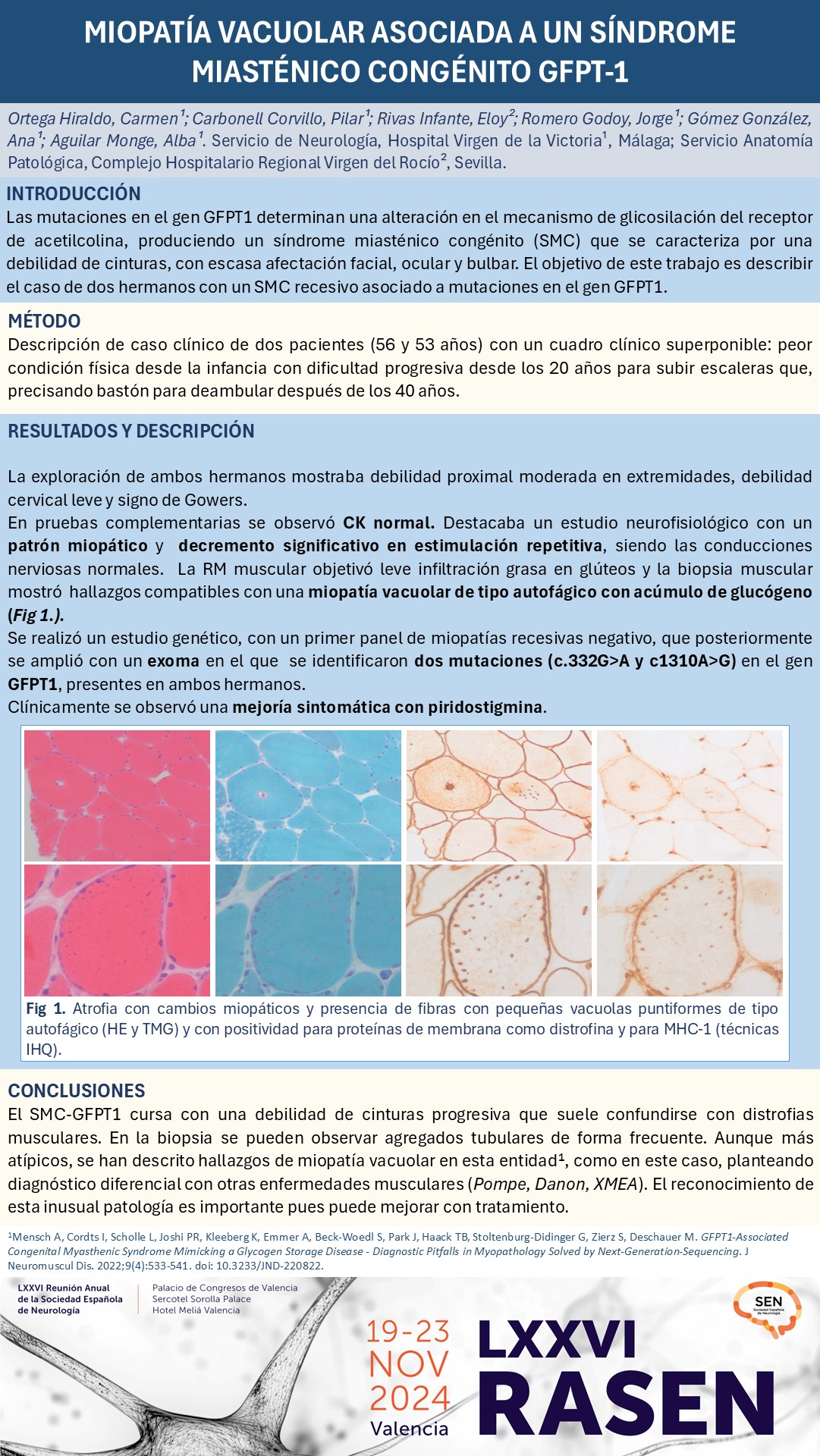
La exploración mostró debilidad proximal moderada en extremidades, debilidad cervical leve y Gowers positivo. La CK fue normal. El estudio neurofisiológico mostró conducciones nerviosas normales, patrón miopático y decremento significativo en la estimulación repetitiva. La RM muscular objetivó leve infiltración grasa en glúteos. La biopsia muscular mostró hallazgos compatibles con una miopatía vacuolar de tipo autofágico con acúmulo de glucógeno. El exoma identificó dos mutaciones (c.332G>A y c1310A>G) en el gen GFPT1, presentes en ambos hermanos. La piridostigmina mejoró los síntomas.
CONCLUSIONES
El SMC-GFPT1 se presenta con una debilidad de cinturas progresiva que suele confundirse con distrofias musculares. Los agregados tubulares son hallazgos frecuentes en la biopsia muscular. Sin embargo, se han descrito hallazgos de miopatía vacuolar en esta entidad, planteando diagnóstico diferencial con otras enfermedades (Pompe, Danon, XMEA). El reconocimiento de esta inusual enfermedad es importante pues puede mejorar con tratamiento.