COMUNICACIÓN POSTER
AUTORES
Jericó Pascual, Ivonne 1; Garcia-Bragado Acin, Federico 2; Ibiricu Yanguas, Asuncion 3; Saenz Bañuelos, Javier 4; Olivé Planas, Montserrat 5
CENTROS
1. Servicio de Neurología. Hospital Virgen del Camino; 2. Servicio: anatomia patológica. Hospital Virgen del Camino; 3. Servicio: Neurofisiología Clínica. Hospital Virgen del Camino; 4. Servicio: Radiología. Hospital Virgen del Camino; 5. Servicio: Unitat neuropatología.. Hospital Universitari de Bellvitge
OBJETIVOS
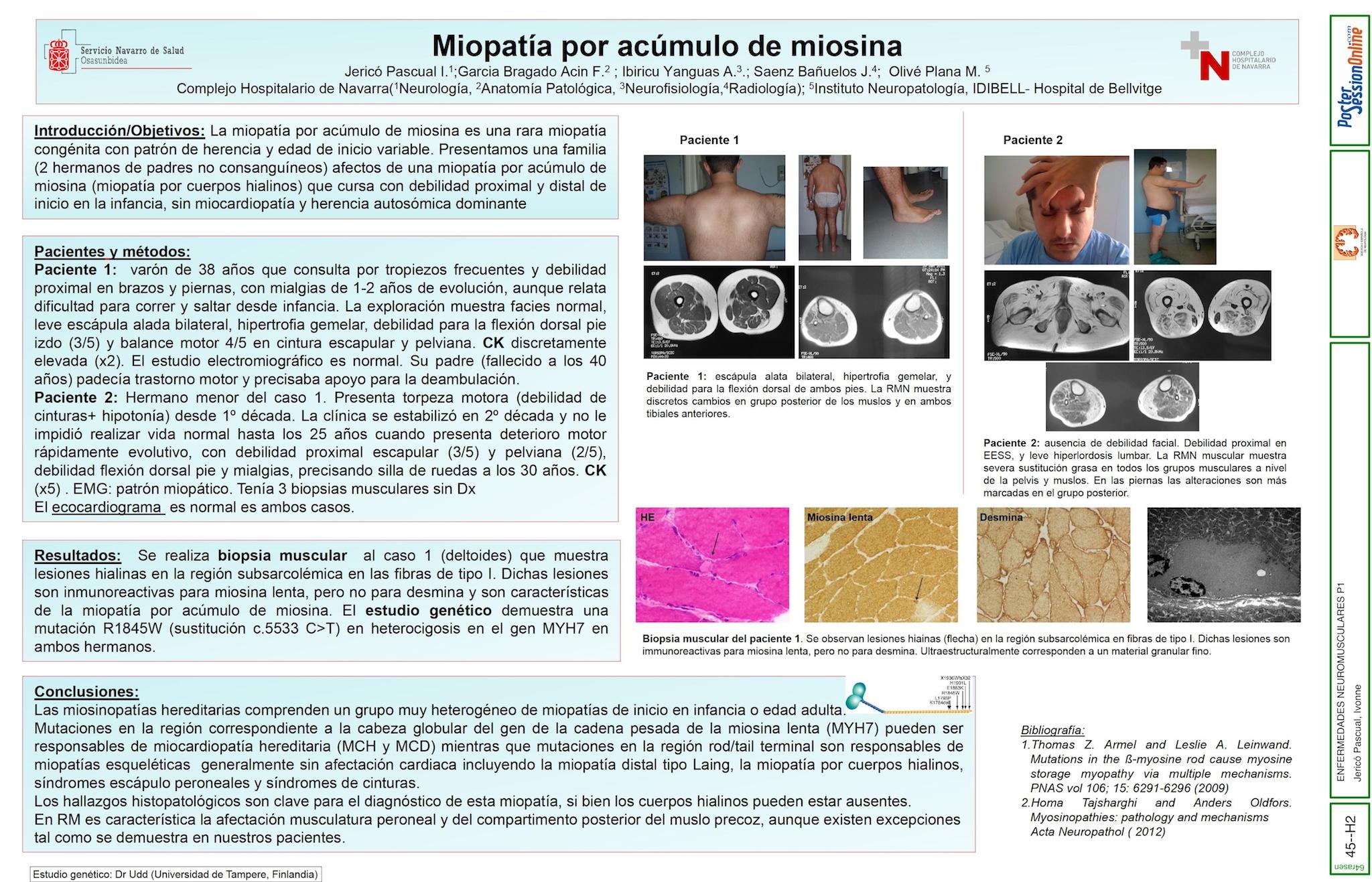
Presentamos una familia (clínica, estudio genético y patrón RM) afecta de miopatía por acúmulo de miosina (miopatía por cuerpos hialinos) con patrón de herencia AD (mutación en el gen MYH-7; CR 14q11.2). Esta miosinopatía hereditaria, con herencia variable y curso lentamente progresivo, presenta 2 fenotipos clínicos predominantes (escapuloperoneal y miopatía cinturas) sin afectación cardiaca.
MATERIAL Y MÉTODOS
(Caso 1)Varón de 38 años con mialgias y debilidad proximal con escápula alada y debilidad asimétrica para la flexión dorsal de pies. (Caso 2) Hermano menor del caso 1 afecto de miopatía de cinturas desde 1º década no progresiva inicialmente, con deterioro rápidamente progresivo a partir de los 25 años, en silla de ruedas desde los 33 años.
RESULTADOS
La miopatía por acúmulo de miosina es una rara entidad, de curso lentamente evolutivo aunque está descrita la posibilidad de deterioro rápidamente progresivo tras varios años de estabilización. Los niveles de CPK son variables. No se acompaña de miocardiopatía. Histológicamente se aprecian depósitos hialinos (miosina lenta +) subsarcolemales exclusivamente en fibras tipo I. El estudio genético en esta familia confirmó la presencia de la mutación en heterocigosidad c.5533C>T(p.R1845W) del gen MYH7.
CONCLUSIONES
Las mutaciones en el gen MYH-7 (Myosin heavy chain-7) son responsables de diferentes síndromes clínicos entre los que se encuentran: miopatía distal tipo III (Gowers-Laing) MPD1, miocardiopatía hipertrófica no compactada, miocardiopatía dilatada y miopatía por cuerpos hialinos. Esta última entidad debe ser considerada en el diagnóstico diferencial de los síndromes escapuloperoneales, aunque hay descritos otros fenotipos.