COMUNICACIÓN POSTER
AUTORES
Marzo Sola, Eugenia 1; Hartung , Hans-Peter 2; Selmaj , Krzysztof 3; Li , David KB 4; Hemmer , Bernhard 5; Freedman , Mark S. 6; Stüve , Olaf 7; Rieckmann , Peter 8; Montalban , Xavier 9; Ziemssen , Tjalf 10; Zhang-Auberson , Lixin 11; Hunter , Brian 11; Rochotte , Erika 11; Wallström , Erik 11; Kappos , Ludwig 12; Gobartt , Ana 13
CENTROS
1. Servicio de Neurología. Hospital San Pedro; 2. Servicio de Neurología. Heinrich Heine University, Dusseldorf, Alemania; 3. Servicio de Neurología. Medical University of Lodz, Lodz, Polonia; 4. Servicio: Radiology and Medicine (Neurology). University of British Columbia, Vancouver, BC, Canadá; 5. Servicio de Neurología. Technical University of Munich, Munich, Alemania; 6. Servicio: -. The Ottawa Hospital Research Institute, University of Ottawa, Ottawa, ON, Canadá; 7. Servicio de Neurología. Technical U. Munich, Alemania; U. Texas SW Medical Center and VA N Texas Health Care Systems, USA; 8. Servicio de Neurología. Sozialstiftung Bamberg Hospital, Bamberg, Alemania; 9. Unidad de Esclerosis múltiple. Hospital Universitari Vall d'Hebron; 10. Servicio de Neurología. Technical University of Dresden, Dresden, Alemania; 11. Servicio: -. Novartis Pharma AG, Basel, Suiza; 12. Servicio de Neurología. University Hospital Basel, Basel, Suiza; 13. Departamento Médico. Novartis Farmacéutica S.A.
OBJETIVOS
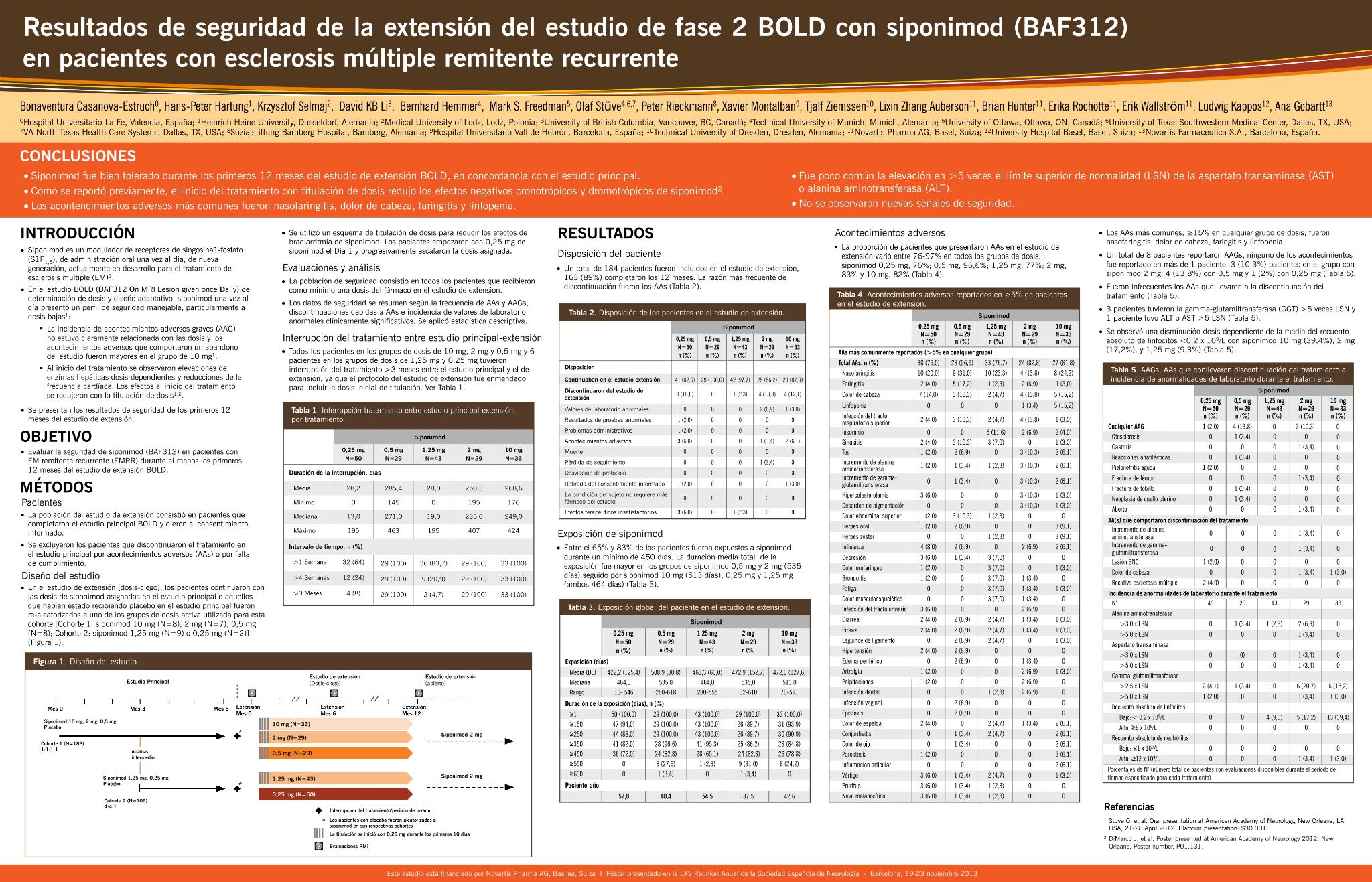
Evaluar la seguridad de siponimod (BAF312) en pacientes con esclerosis múltiple remitente-recurrente durante los primeros 12 meses de extensión del estudio BOLD.
MATERIAL Y MÉTODOS
Los pacientes recibieron las dosis de siponimod asignadas en la fase principal o fueron re-aleatorizados de placebo a siponimod: 33, 29, 43, 29 y 50 pacientes recibieron respectivamente 10, 2, 1,25, 0,5 y 0,25 mg. El 88,6% de los pacientes interrumpió >7días la administración del fármaco entre el estudio principal y la extensión para permitir el ajuste de la dosis de 0,25 mg de siponimod en los primeros 10 días.
RESULTADOS
263/297 pacientes completaron el estudio principal, 184 (62,0%) entraron en la extensión. La exposición a siponimod durante la extensión fue ≥350 días para el 88,6% de los pacientes. Los acontecimientos adversos (AA) más frecuentes (>10%, cualquier grupo) fueron linfopenia, dolor abdominal superior, nasofaringitis, infección del tracto respiratorio superior, faringitis, sinusitis, aumento de ALT o GGT, hipercolesterolemia, cefalea, insomnio, tos, trastorno de la pigmentación. Ocho pacientes (4,3%) notificaron AA graves (AAG): otoesclerosis, gastritis, reacción anafiláctica, pielonefritis aguda, fractura femoral, fractura de tobillo, neoplasia cervical, aborto. La incidencia de AA o AAG no estuvo relacionada con la dosis de siponimod. Los AA llevaron a la interrupción del tratamiento en seis pacientes (3,3%). No se detectaron bradiarritmias sintomáticas en el seguimiento periódico durante el ajuste de dosis inicial.
CONCLUSIONES
Siponimod fue bien tolerado durante los primeros 12 meses de extensión, en concordancia con el estudio principal, y no se observaron nuevos problemas de seguridad.