COMUNICACIÓN POSTER
AUTORES
Lobato Casado, Paula; Morales Casado, Maria Isabel; Segundo Rodríguez, Jose Clemente; Murcia Carretero, Sandra; Vargas Fernández, Celia Cristina; Muñoz Escudero, Francisco; López Ariztegui, Nuria; Marsal Alonso, Carlos
CENTROS
Servicio de Neurología. Hospital Virgen de la Salud
OBJETIVOS
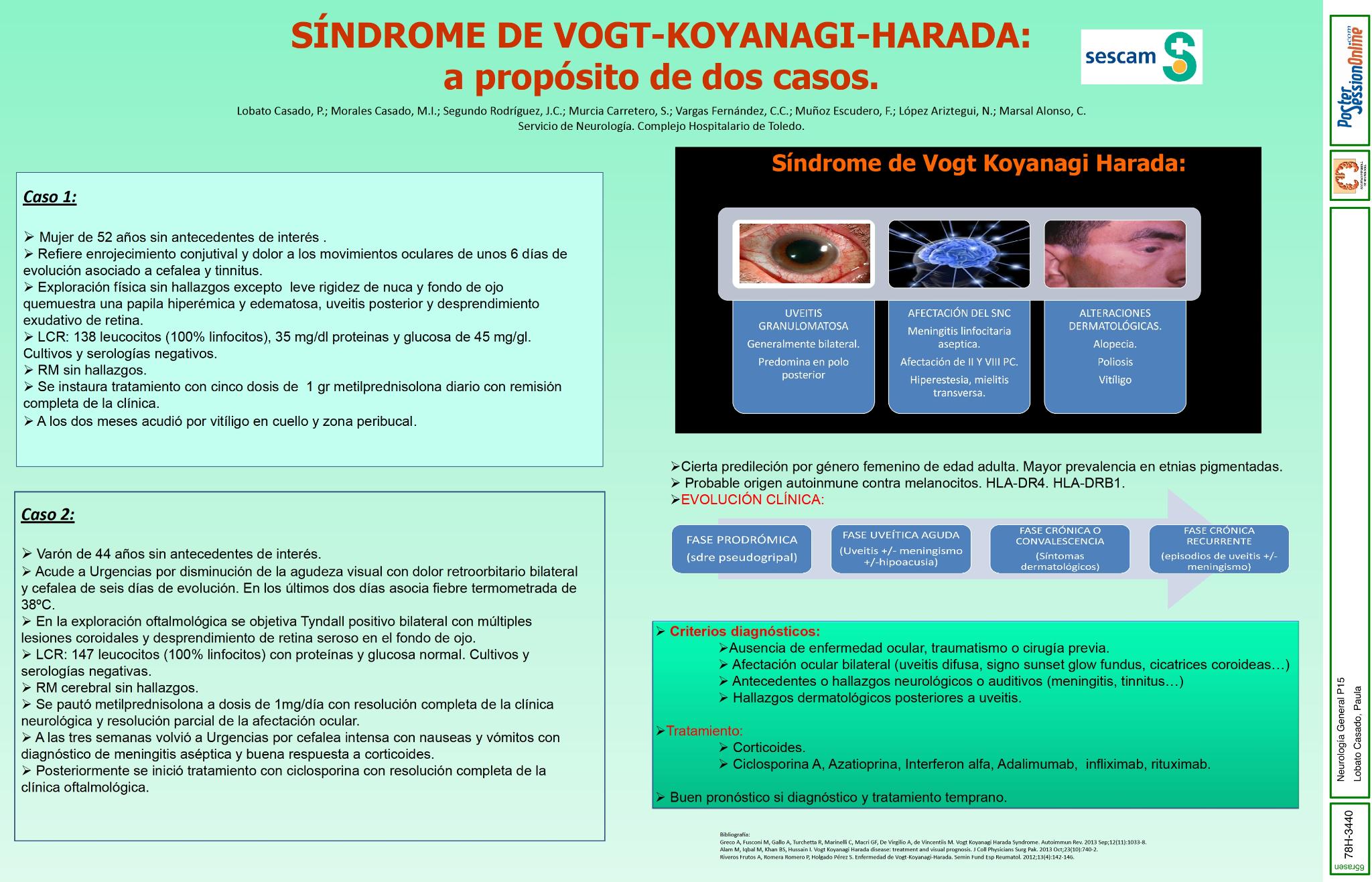
Describir dos casos de síndrome de Vogt-Koyanagi-Harada diagnosticados en nuestro servicio.
MATERIAL Y MÉTODOS
Mujer de 52 años sin antecedentes de interés que consulta por pérdida de visión, inyección conjuntival y dolor con los movimientos oculares asociado a cefalea y tinnitus. El fondo de ojo muestra una papila hiperémica y edematosa y datos de uveitis posterior con daño en el polo posterior de la retina. El LCR muestra 138linfocitos/mm3, con glucosa y proteínas normales, con cultivo negativo. La RM fue normal. Se inició tratamiento con 1gr de metilprednisolona al día durante cinco días con desaparición de la cefalea y el tinnitus y mejoría de la afectación visual. Varón de 44 años sin antecedentes de interés con disminución de la agudeza visual, dolor retroocular, fiebre de 38ºC y cefalea. A la exploración fondo de ojo con lesión en polo posterior de la retina y lesiones coroideas. El LCR muestra 147 linfocitos/mm3 con glucosa y proteínas normales. El cultivo negativo y no se encuentran alteraciones en la RM. Se pautó 1 mg metilprednisolona/día durante 3 días con mejoría clínica.
RESULTADOS
Ambos cuadros podrían englobarse en el síndrome de Vogt-Koyanagi-Harada, patología multisistémica de origen autoinmune caracterizada por uveitis granulomatosa acompañada de alteraciones neurológicas, auditivas y dermatológicas.
CONCLUSIONES
Nos encontramos ante un síndrome poco frecuente (1,5-2% de las uveitis) que siempre habría que tener en cuenta ante un paciente con la sintomatología referida ya que el tratamiento agresivo inicial es esencial para reducir la mortalidad y complicaciones