COMUNICACIÓN POSTER
AUTORES
Pérez Torre, Paula; Escobar Villalba, Alfonso; Estévez Fraga, Carlos; Acebrón Sánchez, Fernando; Martínez Ulloa, Pedro; Guirao Rubio, Carlos; Buisan Catevilla, Francisco Javier; García Barragán, Nuria; Corral Corral, Inigo
CENTROS
Servicio de Neurología. Hospital Ramón y Cajal
OBJETIVOS
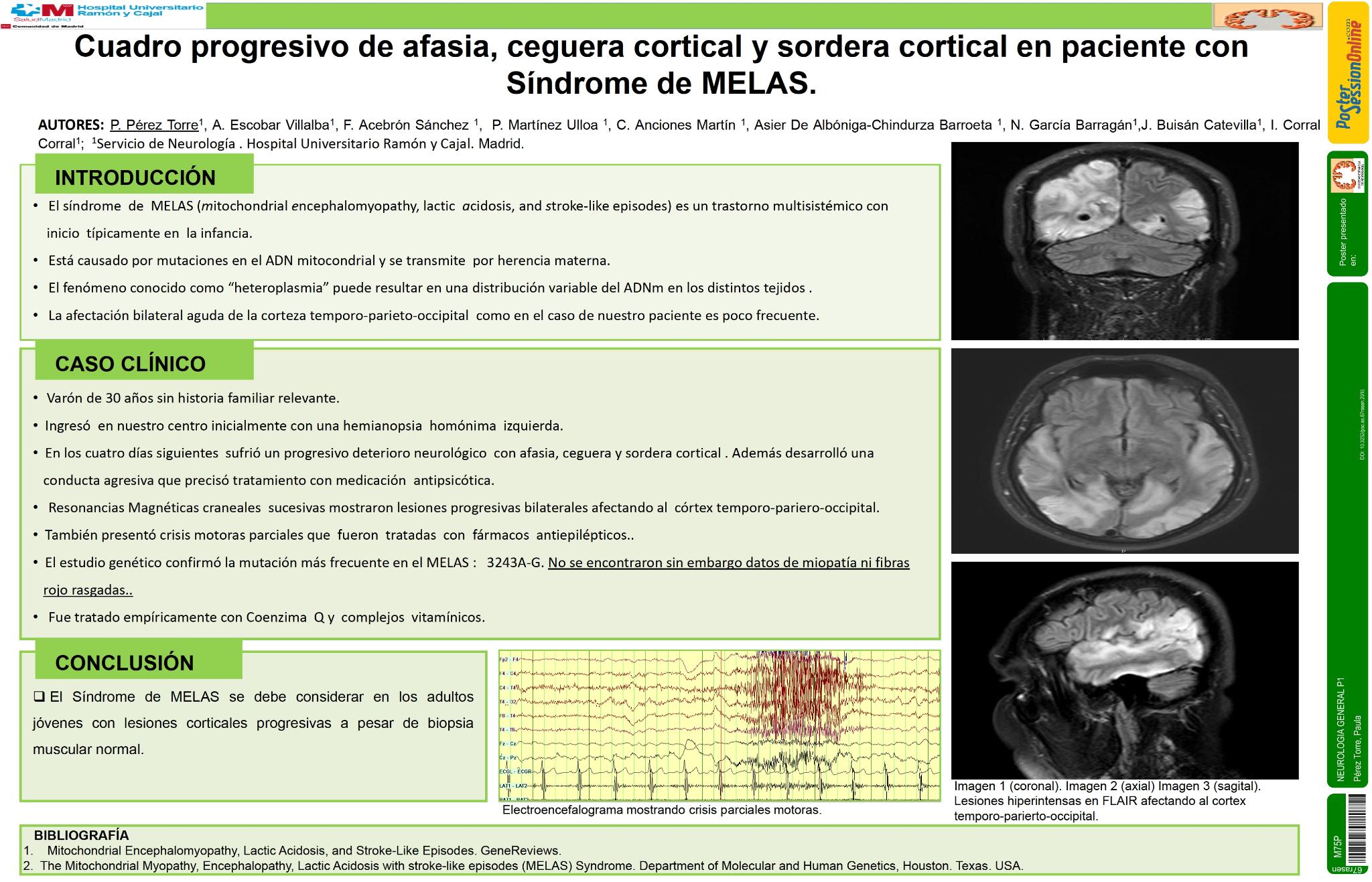
El Síndrome de MELAS (miopatía, encefalopatía, acidosis láctica, episodios stroke-like), frecuentemente presenta alteraciones corticales. La afectación bilateral aguda de la corteza temporo-parieto-occipital es poco frecuente. Presentamos el caso de un paciente con síndrome de MELAS que sufrió episodio progresivo consistente en hemianopsia, afasia, ceguera cortical y sordera cortical.
MATERIAL Y MÉTODOS
Un varón de 30 años sin antecedentes familiares de interés, ingresó en nuestro centro con cuadro de hemianopsia homónima izquierda.El paciente sufrió un rápido deterioro neurológico en las dos semanas siguientes al ingreso,con afasia mixta de predominio sensitivo, crisis epilépticas,ceguera y sordera cortical. Mostraba agresividad,manteniendo contacto con el exterior únicamente por el tacto. Resonancias magnéticas craneales seriadas mostraron lesiones progresivas bilaterales abarcando corteza temporo-parieto-occipital con edema asociado. Desarrolló epilepsia parcial contínua que se manifestó con clonias hemicara derecha y velopalatinas derechas, incluyendo lengua y úvula, que precisaron ajuste de medicación antiepiléptica.
RESULTADOS
La analítica reveló niveles elevados de lactado en sangre y líquido cefalorraquídeo. La biopsia muscular no mostró datos de miopatía o fibras rojo rasgadas. El estudio genético confirmó la mutación 3243A-G. El tratamiento empírico con coenzima Q y complejos vitamínicos resultó en una mejoría significativa a los tres meses.
CONCLUSIONES
El Síndrome de MELAS se debe considerar en los adultos jóvenes con lesiones corticales progresivas a pesar biopsia muscular normal.