COMUNICACIÓN POSTER
AUTORES
San Miguel Oroz, Mikel 1; Torné Hernández, Laura 2; Pagola Elorz, Inmaculada 2; Tellechea Aramburo, Paula 2; Pasalodos , Sara 3; Salgado , Josefa 3; Alonso , Angel 3; Mendióroz Iriarte, Maite 2; Jericó Pascual, Ivonne 2
CENTROS
1. Servicio de Neurología. Hospital de Navarra; 2. Servicio de Neurología. Complejo Hospitalario de Navarra; 3. Servicio: Medicina Genómica. Complejo Hospitalario de Navarra
OBJETIVOS
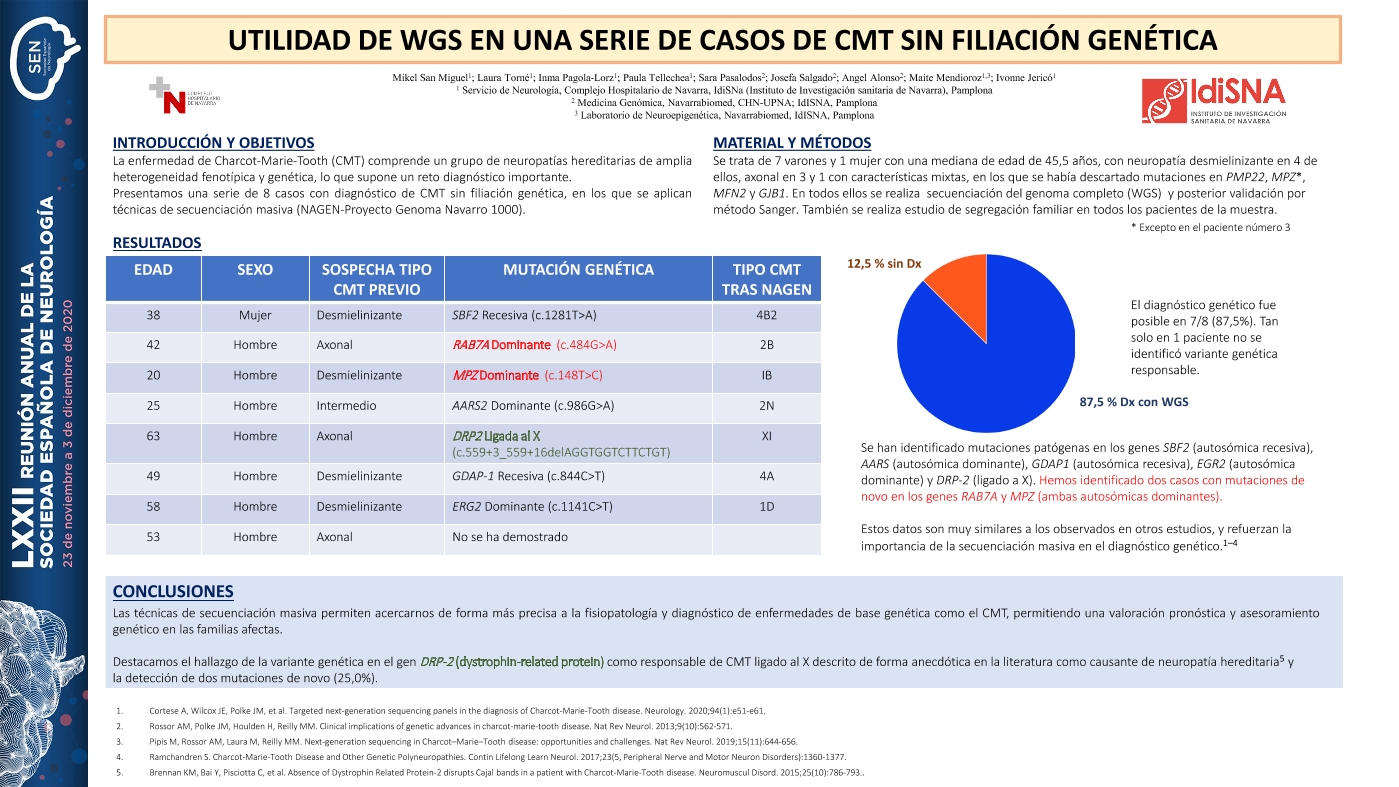
La enfermedad de Charcot-Marie-Tooth (CMT) comprende un grupo de neuropatías hereditarias de amplia heterogeneidad fenotípica y genética, lo que supone un reto diagnóstico importante. Presentamos una serie de 9 casos con diagnóstico de CMT sin filiación genética, en los que se aplican técnicas de secuenciación masiva.
MATERIAL Y MÉTODOS
Se trata de 8 varones y 1 mujer con una mediana de edad de 38 años, con neuropatía desmielinizante en 4 de ellos, axonal en 4 y 1 con características mixtas, en los que se había descartado mutaciones en PMP22, MPZ, MFN2 y GJB1. En todos ellos se realiza secuenciación del genoma completo (WGS) a través del proyecto NAGEN-1000 y posterior validación por método Sanger.
RESULTADOS
El diagnóstico genético fue posible en 7/9 (77,8%). Tan solo en 2 pacientes no se identificó variante genética responsable. Se han identificado mutaciones patógenas en los genes SBF2 (autosómica recesiva), AARS (autosómica dominante), GDAP1 (autosómica recesiva), EGR2 (autosómica dominante) y DRP-2 (ligado a X). Hemos identificado dos casos con mutaciones de novo en los genes RAB7A y MPZ (ambas autosómicas dominantes)
CONCLUSIONES
Las técnicas de secuenciación masiva permiten acercarnos de forma más precisa a la fisiopatología y diagnóstico de enfermedades de base genética como el CMT, permitiendo una valoración pronóstica y asesoramiento genético en las familias afectas. Destacamos el hallazgo de la variante genética en el gen DRP-2 (dystrophin-related protein) como responsable de CMT ligado al X descrito de forma anecdótica en la literatura como causante de neuropatía hereditaria y la detección de dos mutaciones de novo (22,2%)