COMUNICACIÓN POSTER
AUTORES
Benetó Andrés, Helena 1; Villanueva Haba, Vicente 2; Garcés Sánchez, Mercedes 2; null, Kevin 2
CENTROS
1. Servicio de Neurología. Hospital General de Castelló; 2. Servicio de Neurología. Hospital Universitari i Politècnic La Fe
OBJETIVOS
Las variantes patogénicas dominantes en el gen SCN1A condicionan una pérdida de función por haploinsuficiencia de los canales de sodio dependientes de voltaje en las neuronas inhibitorias y son una de las causas más frecuentes de epilepsia monogénica. Su espectro clínico es variable, pudiéndose presentar como síndrome de Dravet en su forma más grave. Presentamos un estudio observacional, transversal, cuyo objetivo fue definir las características de los pacientes con síndrome de Dravet en seguimiento por una unidad especializada en epilepsia refractaria.
MATERIAL Y MÉTODOS
Se recogieron datos de 15 pacientes con síndrome de Dravet confirmado genéticamente a través de la historia clínica y de una entrevista telefónica con el cuidador principal en la que se evaluaron la epilepsia, la cognición, la conducta y los trastornos del movimiento utilizando, para guiar la entrevista, la escala MDS-UPDRS y el cuestionario de Conners y, posteriormente, se realizó un análisis descriptivo de los mismos.
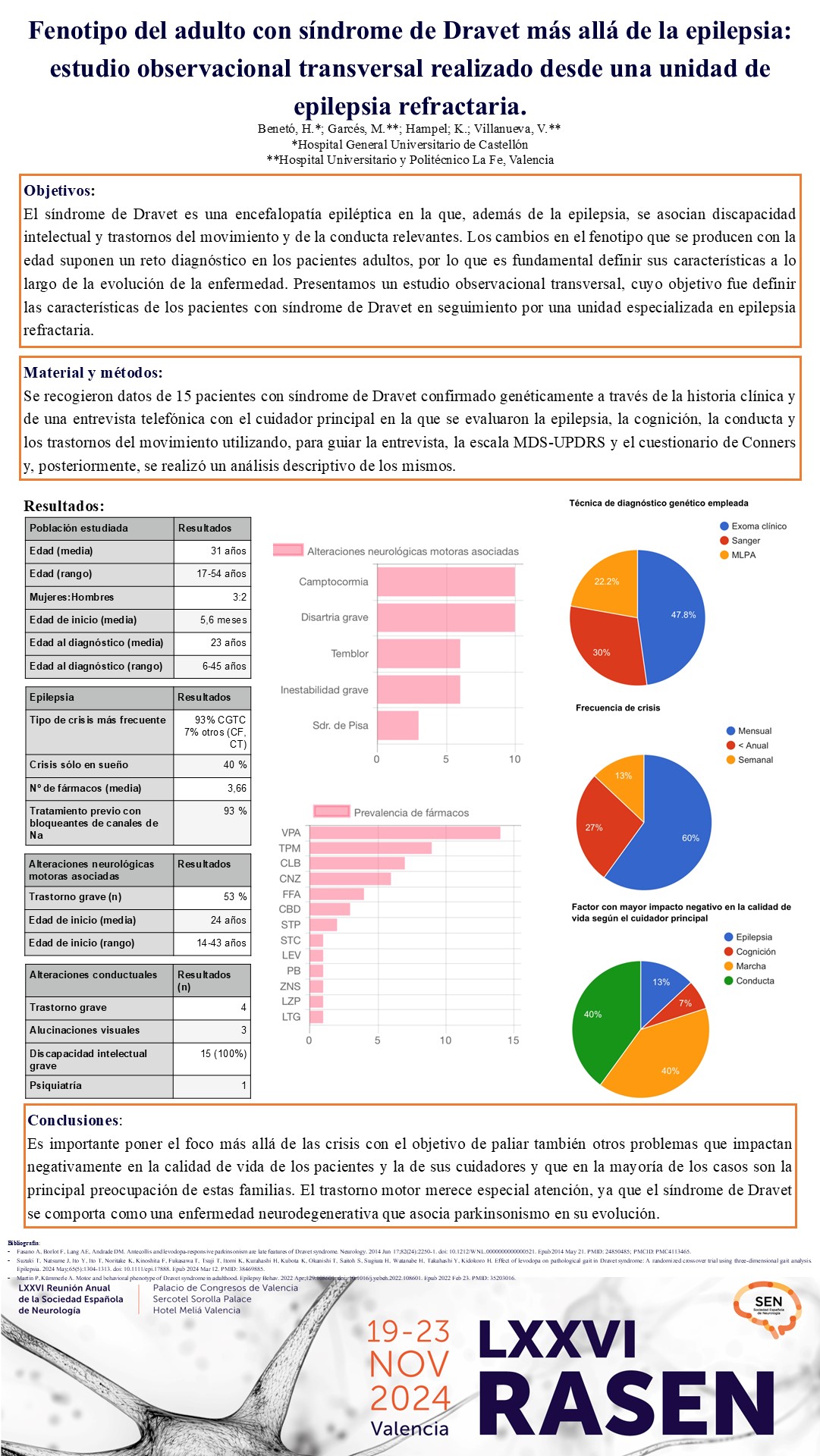
RESULTADOS
Se describen datos sobre la epilepsia (grado de control y tratamiento), los trastornos de conducta/psiquiátricos y del movimiento, así como de la percepción de calidad de vida por parte de la familia en la población estudiada.
CONCLUSIONES
Conocer la historia natural del síndrome Dravet y el fenotipo clínico con el que se presenta en las diferentes etapas de la vida es fundamental para poner fin al retraso diagnóstico en este grupo de pacientes y de esta manera poder plantear un manejo personalizado de forma precoz, evitándose fármacos deletéreos y facilitándose el acceso a tratamientos específicos e incluso a ensayos clínicos.